Más sobre ciencia de datos en: cienciadedatos.net
- Regresión lineal con Python
- Regresión lineal múltiple con Python
- Regresión logística con Python
- Regularización Ridge, Lasso y Elastic Net con Python
- Machine learning con Python y Scikitlearn
- Árboles de decisión con Python: regresión y clasificación
- Random Forest con Python y Scikit-learn
- Gradient Boosting con Python y Scikit-learn
- Gradient Boosting probabilístico con Python
- Máquinas de Vector Soporte (SVM)
- Redes neuronales con Python
- Análisis de componentes principales PCA
- Clustering
- Detección de anomalías con PCA
- Detección de anomalías con autoencoders
- Detección de anomalías con Gaussian Mixture Models
- Detección de anomalías con Isolation Forest
- Análisis de texto (text mining) con Python
- Reglas de asociación
- Análisis Tweets con Python
Introducción¶
Un modelo Random Forest está formado por múltiples árboles de decisión individuales. Cada uno de estos árboles es entrenado con una muestra ligeramente diferente de los datos de entrenamiento, generada mediante una técnica conocida como bootstrapping. Para realizar predicciones sobre nuevas observaciones, se combinan las predicciones de todos los árboles que conforman el modelo.
Muchos métodos predictivos generan modelos globales en los que una única ecuación se aplica a todo el espacio muestral. Cuando el caso de uso implica múltiples predictores, que interaccionan entre ellos de forma compleja y no lineal, es muy difícil encontrar un único modelo global que sea capaz de reflejar la relación entre las variables. Los métodos estadísticos y de machine learning basados en árboles engloban a un conjunto de técnicas supervisadas no paramétricas que consiguen segmentar el espacio de los predictores en regiones simples, dentro de las cuales es más sencillo manejar las interacciones. Es esta característica la que les proporciona gran parte de su potencial.
Los métodos basados en árboles han ganado reconocimiento como una referencia en el ámbito de la predicción debido a los excelentes resultados que ofrecen en una amplia gama de problemas. En este documento se explora cómo se construyen y utilizan los modelos Random Forest.
✎ Nota
Es importante conocer el funcionamiento de los árboles de decisión para poder comprender los modelos Random Forest. También es recomendable conocer los modelos Gradient Boosting, que comparten muchas carácteristicas con Random Forest.Ventajas
Son capaces de seleccionar predictores más relevantes de forma automática.
Pueden aplicarse a problemas de regresión y clasificación.
Los árboles pueden, en teoría, manejar tanto predictores numéricos como categóricos sin tener que crear variables dummy o one-hot-encoding. En la práctica, esto depende de la implementación del algoritmo que tenga cada librería.
Al ser métodos no paramétricos, no requieren que los datos sigan una distribución específica.
Por lo general, necesitan menos limpieza y preprocesamiento de datos en comparación con otros métodos de aprendizaje estadístico. Por ejemplo, no requieren estandarización.
Son menos susceptibles a ser influenciados por valores atípicos (outliers).
Si el valor de un predictor no está disponible para una observación, aún se puede realizar una predicción utilizando las observaciones del último nodo alcanzado. La precisión de la predicción se verá reducida pero al menos podrá obtenerse.
Son muy útiles en la exploración de datos, permiten identificar de forma rápida y eficiente las variables (predictores) más importantes.
Gracias al Out-of-Bag Error, es posible estimar el error de validación sin recurrir a estrategias computacionalmente costosas como la validación cruzada. Esto no aplica en el caso de series temporales.
Son adecuados para conjuntos de datos con un gran número de observaciones, demostrando una buena escalabilidad.
Desventajas
La combinación de múltiples árboles reduce la capacidad de interpretación en comparación con modelos basados en un solo árbol.
Al tratar con predictores continuos, se puede perder parte de la información al categorizarlos durante la división de los nodos.
La técnica de recursive binary splitting utilizada para crear las ramificaciones de los árboles puede favorecer a los predictores continuos o cualitativos con muchos niveles, ya que tienen una mayor probabilidad de contener un punto de corte óptimo por azar.
Tal y como se describe más adelante, la creación de las ramificaciones de los árboles se consigue mediante el algoritmo de recursive binary splitting. Este algoritmo identifica y evalúa las posibles divisiones de cada predictor acorde a una determinada medida (RSS, Gini, entropía…). Los predictores continuos o predictores cualitativos con muchos niveles tienen mayor probabilidad de contener, solo por azar, algún punto de corte óptimo, por lo que suelen verse favorecidos en la creación de los árboles.
No son capaces de extrapolar fuera del rango observado en los datos de entrenamiento.
Random Forest en Python
Existen múltiples implementaciones de modelos Random Forest en Python, siendo una de las más utilizadas es la disponible en scikit-learn. Aunque es menos conocido, las principales librerías de Gradient Boosting como LightGBM y XGBoost también pueden configurarse para crear modelos Random Forest. Todas ellas están muy optimizadas y se utilizan de forma similar, sin embargo, presentan diferencias en su implementación que pueden conducir a resultados distintos. En scikit-learn, es necesario aplicar one-hot-encoding a los predictores categóricos, cosa que no es necesaria en LightGBM y XGBoost. Esto incide directamente en la estructura de los árboles generados y, por consiguiente, en los resultados predictivos del modelo y en la importancia atribuida a los predictores (consultar detalles más adelante).
✎ Nota
A partir de la version1.4, los modelos RandomForest de scikit-learn soportan valores ausentes.
Random Forest¶
Un modelo Random Forest está compuesto por un conjunto (ensemble) de árboles de decisión individuales. Cada uno de estos árboles es entrenado con una muestra aleatoria extraída de los datos de entrenamiento originales mediante bootstrapping). Esto implica que cada árbol se entrena con un conjunto de datos ligeramente diferente. En cada árbol individual, las observaciones se distribuyen a través de bifurcaciones (nodos), dando forma a la estructura del árbol hasta llegar a un nodo terminal. La predicción de una nueva observación se obtiene al agregar las predicciones de todos los árboles individuales que conforman el modelo.
Para comprender cómo funcionan los modelos Random Forest es fundamental familiarizarse primero con los conceptos de ensemble y bagging.
Métodos de ensemble¶
Todos los modelos de aprendizaje estadístico y machine learning enfrentan el desafío del equilibrio entre sesgo y varianza.
El término "sesgo" (bias) se refiere a cuánto se desvían en promedio las predicciones de un modelo con respecto a los valores reales. Refleja la habilidad del modelo para capturar la verdadera relación entre los predictores y la variable de respuesta. Por ejemplo, si la relación sigue un patrón no lineal, un modelo de regresión lineal, independientemente de cuántos datos se disponga, no podrá modelar adecuadamente la relación y tendrá un sesgo alto.
Por otro lado, el término "varianza" hace referencia a cuánto cambia el modelo en función de los datos utilizados en su entrenamiento. Idealmente, un modelo no debería cambiar demasiado ante pequeñas variaciones en los datos de entrenamiento. Si esto ocurre, indica que el modelo está memorizando los datos en lugar de aprender la verdadera relación entre los predictores y la variable de respuesta. Por ejemplo, un modelo de árbol con muchos nodos tiende a cambiar su estructura incluso con pequeñas variaciones en los datos de entrenamiento, lo que indica que tiene alta varianza.
Conforme aumenta la complejidad de un modelo, este dispone una mayor flexibilidad para adaptarse a las observaciones, lo que conlleva una reducción del sesgo y una mejora en su capacidad predictiva. Sin embargo, una vez alcanzado un cierto nivel de complejidad, surge el problema del sobreajuste (overfitting). Este fenómeno se presenta cuando el modelo se ajusta tanto a los datos de entrenamiento que es incapaz de predecir correctamente nuevas observaciones. El modelo óptimo es aquel que logra encontrar un equilibrio adecuado entre sesgo y varianza.
¿Cómo se controlan el bias y varianza en los modelos basados en árboles? Por lo general, los árboles pequeños, con pocas ramificaciones, tienden a tener poca varianza pero pueden no captar de manera precisa la relación entre las variables, lo que se traduce en un alto sesgo. Por otro lado, los árboles grandes se ajustan mucho a los datos de entrenamiento, lo que reduce el sesgo pero incrementa la varianza. Una forma de solucionar este problema son los métodos de ensemble.
Los métodos de ensemble combinan múltiples modelos en uno nuevo con el objetivo de lograr un equilibro entre bias y varianza, consiguiendo así mejores predicciones que cualquiera de los modelos individuales originales. Dos de los tipos de ensemble más utilizados son:
Bagging: Se ajustan múltiples modelos, cada uno con un subconjunto distinto de los datos de entrenamiento. Para predecir, todos los modelos que forman el agregado participan aportando su predicción. Como valor final, se toma la media de todas las predicciones (variables continuas) o la clase más frecuente (variables categóricas). Los modelos Random Forest están dentro de esta categoría.
Boosting: Se ajustan secuencialmente múltiples modelos sencillos, llamados weak learners, de forma que cada modelo aprende de los errores del anterior. Como valor final, al igual que en bagging, se toma la media de todas las predicciones (variables continuas) o la clase más frecuente (variables cualitativas). Tres de los métodos de boosting más empleados son AdaBoost, Gradient Boosting y Stochastic Gradient Boosting.
Aunque el objetivo final es el mismo, lograr un balance óptimo entre bias y varianza, existen dos diferencias importantes:
Forma en que consiguen reducir el error total. El error total de un modelo puede descomponerse como $bias + varianza + \epsilon$. En bagging, se emplean modelos con muy poco bias pero mucha varianza, agregándolos se consigue reducir la varianza sin apenas inflar el bias. En boosting, se emplean modelos con muy poca varianza pero mucho bias, ajustando secuencialmente los modelos se reduce el bias. Por lo tanto, cada una de las estrategias reduce una parte del error total.
Forma en que se introducen variaciones en los modelos que forman el ensemble. En bagging, cada modelo es distinto del resto porque cada uno se entrena con una muestra distinta obtenida mediante bootstrapping). En boosting, los modelos se ajustan secuencialmente y la importancia (peso) de las observaciones va cambiando en cada iteración, dando lugar a diferentes ajustes.
La clave para que los métodos de ensemble consigan mejores resultados que cualquiera de sus modelos individuales es que, los modelos que los forman, sean lo más diversos posibles (sus errores no estén correlacionados). Una analogía que refleja este concepto es la siguiente: supóngase un juego como el trivial en el que los equipos tienen que acertar preguntas sobre temáticas diversas. Un equipo formado por muchos jugadores, cada uno experto en un tema distinto, tendrá más posibilidades de ganar que un equipo formado por jugadores expertos en un único tema o por un único jugador que sepa un poco de todos los temas.
A continuación, se describe con más detalle la estrategia de bagging, sobre la que se fundamenta el modelo Random Forest.
Bagging¶
El término bagging es el diminutivo de bootstrap aggregation, y hace referencia al empleo del muestreo repetido con reposición bootstrapping) con el fin de reducir la varianza de algunos modelos de aprendizaje estadístico, entre ellos los basados en árboles.
Dadas $n$ muestras de observaciones independientes $Z_1$, ..., $Z_n$, cada una con varianza $\sigma^2$, la varianza de la media de las observaciones $\overline{Z}$ es $\sigma^2/n$. En otras palabras, promediando un conjunto de observaciones se reduce la varianza. Basándose en esta idea, una forma de reducir la varianza y aumentar la precisión de un método predictivo es obtener múltiples muestras de la población, ajustar un modelo distinto con cada una de ellas, y hacer la media (la moda en el caso de variables cualitativas) de las predicciones resultantes. Como en la práctica no se suele tener acceso a múltiples muestras, se puede simular el proceso recurriendo a bootstrapping), generando así pseudo-muestras con los que ajustar diferentes modelos y después agregarlos. A este proceso se le conoce como bagging y es aplicable a una gran variedad de métodos de regresión.
En el caso particular de los árboles de decisión, dada su naturaleza de bajo bias y alta varianza, bagging ha demostrado tener muy buenos resultados. La forma de aplicarlo es:
Generar $B$ pseudo-training sets mediante bootstrapping a partir de la muestra de entrenamiento original.
Entrenar un árbol con cada una de las $B$ muestras del paso 1. Cada árbol se crea sin apenas restricciones y no se somete a pruning, por lo que tiene varianza alta pero poco bias. En la mayoría de casos, la única regla de parada es el número mínimo de observaciones que deben tener los nodos terminales. El valor óptimo de este hiperparámetro puede obtenerse comparando el out of bag error o por validación cruzada.
Para cada nueva observación, obtener la predicción de cada uno de los $B$ árboles. El valor final de la predicción se obtiene como la media de las $B$ predicciones en el caso de variables cuantitativas y como la clase predicha más frecuente (moda) para variables cualitativas.
En el proceso de bagging, el número de árboles creados no es un hiperparámetro crítico en cuanto a que, por mucho que se incremente el número, no se aumenta el riesgo de overfitting. Alcanzado un determinado número de árboles, la reducción de test error se estabiliza. A pesar de ello, cada árbol ocupa memoria, por lo que no conviene almacenar más de los necesarios.
Entrenamiento de Random Forest¶
El algoritmo de Random Forest es una modificación del proceso de bagging que consigue mejorar los resultados gracias a que decorrelaciona aún más los árboles generados en el proceso.
Recordando el apartado anterior, los beneficios de bagging se basan en el hecho de que, promediando un conjunto de modelos, se consigue reducir la varianza. Esto es cierto siempre y cuando los modelos agregados no estén correlacionados. Si la correlación es alta, la reducción de varianza que se puede lograr es pequeña.
Supóngase un set de datos en el que hay un predictor muy influyente, junto con otros moderadamente influyentes. En este escenario, todos o casi todos los árboles creados en el proceso de bagging estarán dominados por el mismo predictor y serán muy parecidos entre ellos. Como consecuencia de la alta correlación entre los árboles, el proceso de bagging apenas conseguirá disminuir la varianza y, por lo tanto, tampoco mejorar el modelo. Random forest evita este problema haciendo una selección aleatoria de $m$ predictores antes de evaluar cada división. De esta forma, un promedio de $(p-m)/p$ divisiones no contemplarán el predictor influyente, permitiendo que otros predictores puedan ser seleccionados. Añadiendo este paso extra se consigue decorrelacionar los árboles todavía más, con lo que su agregación consigue una mayor reducción de la varianza.
Los métodos de random forest y bagging siguen el mismo algoritmo con la única diferencia de que, en random forest, antes de cada división, se seleccionan aleatoriamente $m$ predictores. La diferencia en el resultado dependerá del valor $m$ escogido. Si $m=p$ los resultados de random forest y bagging son equivalentes. Algunas recomendaciones son:
La raíz cuadrada del número total de predictores para problemas de clasificación. $m \approx \sqrt{p}$
Un tercio del número de predictores para problemas de regresión. $m \approx \frac{p}{3}$
Si los predictores están muy correlacionados, valores pequeños de $m$ consiguen mejores resultados.
Sin embargo, la mejor forma para encontrar el valor óptimo de $m$ es evaluar el out-of-bag-error o recurrir a validación cruzada.
Al igual que ocurre con bagging, random forest no sufre problemas de overfit por aumentar el número de árboles creados en el proceso. Alcanzado un determinado número, la reducción del error de test se estabiliza.
Predicción de Random Forest¶
La predicción de un modelo Random Forest es la media de las predicciones de todos los árboles que lo forman.
Supóngase que se dispone de 10 observaciones, cada una con un valor de variable respuesta $Y$ y unos predictores $X$.
| id | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| Y | 10 | 18 | 24 | 8 | 2 | 9 | 16 | 10 | 20 | 14 |
| X | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
La siguiente imagen muestra cómo sería la predicción del modelo para una nueva observación. En cada árbol, el camino hasta llegar al nodo final está resaltado. En cada nodo terminal se detalla el índice de las observaciones de entrenamiento que forman parte.
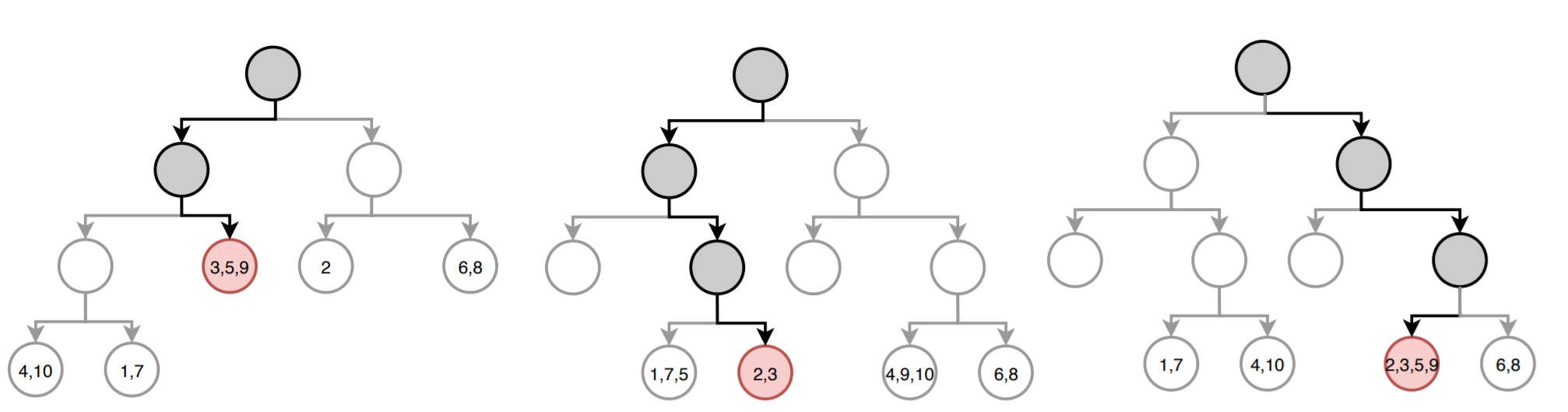
El valor predicho por cada árbol es la media de la variable respuesta $Y$ en el nodo terminal. Acorde a la imagen, las predicciones de cada uno de los tres árboles (de izquierda a derecha) es:
$$\hat{y}_{arbol_1} = \frac{24 + 2 + 20}{3} = 15.33333$$$$\hat{y}_{arbol_2} = \frac{18 + 24}{2} = 21$$$$\hat{y}_{arbol_3} = \frac{18 + 24 + 2 + 20}{4} = 16$$La predicción final del modelo es la media de todas las predicciones individuales:
$$\hat{\mu} = \frac{15.33333+21+16}{3} = 17.4$$Aunque la anterior es la forma más común de obtener las predicciones de un modelo Random Forest, existe otra aproximación. La predicción de un árbol de regresión puede verse como una variante de vecinos cercanos en la que, solo las observaciones que forman parte del mismo nodo terminal que la observación predicha, tienen influencia. Siguiendo esta aproximación, la predicción del árbol se define como la media ponderada de todas las observaciones de entrenamiento, donde el peso de cada observación depende únicamente de si forma parte o no del mismo nodo terminal. Para Random Forest esto equivale a la media ponderada de todas las observaciones, empleando como pesos $\textbf{w}$ la media de los vectores de pesos de todos los árboles.
Acorde a la imagen anterior, el vector de pesos para cada uno de los tres árboles (de izquierda a derecha) es:
$$\textbf{w}_{arbol_1} = (0, 0, \frac{1}{3}, 0, \frac{1}{3}, 0, 0, 0, \frac{1}{3}, 0)$$$$\textbf{w}_{arbol_2} = (0, \frac{1}{2}, \frac{1}{2}, 0, 0, 0, 0, 0, 0, 0)$$$$\textbf{w}_{arbol_3} = (0, \frac{1}{4}, \frac{1}{4}, 0, \frac{1}{4}, 0, 0, 0, \frac{1}{4}, 0)$$La media de todos los vectores de pesos es:
$$\overline{\textbf{w}} = \frac{1}{3}(\textbf{w}_{arbol_1} + \textbf{w}_{arbol_2} + \textbf{w}_{arbol_3}) = \\ (0, \frac{1}{4}, \frac{13}{36}, 0, \frac{7}{36}, 0, 0, 0, \frac{7}{36}, 0)$$Una vez obtenido el vector de pesos promedio, se puede calcular la predicción con la media ponderada de todas las observaciones de entrenamiento:
$$\hat{\mu} = \sum^n_{i =1} \overline{\textbf{w}}_i Y_i$$$$\hat{\mu} = (0\times10) + (\frac{1}{4}\times18) + (\frac{13}{36}\times24) + (0\times8) + (\frac{1}{4}\times2) + \\ (0\times9) + (0\times16) + (0\times10) + (\frac{1}{4}\times20) + (0\times14) = 17.4$$Out-of-Bag Error¶
Dada la naturaleza del proceso de bagging, resulta posible estimar el error de test sin necesidad de recurrir a métodos de validación cruzada (cross-validation). El hecho de que los árboles se ajusten empleando muestras generadas por bootstrapping conlleva que, en promedio, cada ajuste use solo aproximadamente dos tercios de las observaciones originales. Al tercio restante se le llama out-of-bag (OOB).
Si para cada árbol ajustado en el proceso de bagging se registran las observaciones empleadas, se puede predecir la respuesta de la observación $i$ haciendo uso de aquellos árboles en los que esa observación ha sido excluida y promediándolos (la moda en el caso de los árboles de clasificación). Siguiendo este proceso, se pueden obtener las predicciones para las $n$ observaciones y con ellas calcular el OOB-mean square error (para regresión) o el OOB-classification error (para árboles de clasificación). Como la variable respuesta de cada observación se predice empleando únicamente los árboles en cuyo ajuste no participó dicha observación, el OOB-error sirve como estimación del error de test. De hecho, si el número de árboles es suficientemente alto, el OOB-error es prácticamente equivalente al leave-one-out cross-validation error.
Esta es una ventaja añadida de los métodos de bagging, y por lo tanto de Random Forest ya que evita tener que recurrir al proceso de validación cruzada (computacionalmente costoso) para la optimización de los hiperparámetros.
Dos limitaciones en el uso Out-of-Bag Error:
El Out-of-Bag Error no es adecuado cuando las observaciones tienen una relación temporal (series temporales). Como la selección de las observaciones que participan en cada entrenamiento es aleatoria, no respetan el orden temporal y se estaría introduciendo información a futuro.
El preprocesado de los datos de entrenamiento se hace de forma conjunta, por lo que las observaciones out-of-bag pueden sufrir data leakage). De ser así, las estimaciones del OOB-error son demasiado optimistas.
En un muestreo por bootstrapping, si el tamaño de los datos de entrenamiento es $n$, cada observación tiene una probabilidad de ser elegida de $\frac{1}{n}$. Por lo tanto, la probabilidad de no ser elegida en todo el proceso es de $(1-1/n)n$, lo que converge en $1/\epsilon$, que es aproximadamente un tercio.
Importancia de los predictores¶
Si bien es cierto que el proceso de bagging (Random Forest) consigue mejorar la capacidad predictiva en comparación a los modelos basados en un único árbol, esto tiene un coste asociado, la interpretabilidad del modelo se reduce. Al tratarse de una combinación de múltiples árboles, no es posible obtener una representación gráfica sencilla del modelo y no es inmediato identificar de forma visual que predictores son más importantes. Sin embargo, se han desarrollado nuevas estrategias para cuantificar la importancia de los predictores que hacen de los modelos de bagging (Random Forest) una herramienta muy potente, no solo para predecir, sino también para el análisis exploratorio. Dos de estas medidas son: importancia por permutación e impureza de nodos.
Importancia por permutación
Identifica la influencia que tiene cada predictor sobre una determinada métrica de evaluación del modelo (estimada por out-of-bag error o validación cruzada). El valor asociado con cada predictor se obtiene de la siguiente forma:
Crear el conjunto de árboles que forman el modelo.
Calcular una determinada métrica de error (mse, classification error, ...). Este es el valor de referencia ($error_0$).
Para cada predictor $j$:
Permutar en todos los árboles del modelo los valores del predictor $j$ manteniendo el resto constante.
Recalcular la métrica tras la permutación, llámese ($error_j$).
Calcular el incremento en la métrica debido a la permutación del predictor $j$.
Si el predictor permutado estaba contribuyendo al modelo, es de esperar que el modelo aumente su error, ya que se pierde la información que proporcionaba esa variable. El porcentaje en que se incrementa el error debido a la permutación del predictor $j$ puede interpretarse como la influencia que tiene $j$ sobre el modelo. Algo que suele llevar a confusiones es el hecho de que este incremento puede resultar negativo. Si la variable no contribuye al modelo, es posible que, al reorganizarla aleatoriamente, solo por azar, se consiga mejorar ligeramente el modelo, por lo que $(error_j - error_0)$ es negativo. A modo general, se puede considerar que estas variables tiene una importancia próxima a cero.
Aunque esta estrategia suele ser la más recomendado, cabe tomar algunas precauciones en su interpretación. Lo que cuantifican es la influencia que tienen los predictores sobre el modelo, no su relación con la variable respuesta. ¿Por qué es esto tan importante? Supóngase un escenario en el que se emplea esta estrategia con la finalidad de identificar qué predictores están relacionados con el peso de una persona, y que dos de los predictores son: el índice de masa corporal (IMC) y la altura. Como IMC y altura están muy correlacionados entre sí (la información que aportan es redundante), cuando se permute uno de ellos, el impacto en el modelo será mínimo, ya que el otro aporta la misma información. Como resultado, estos predictores aparecerán como poco influyentes aun cuando realmente están muy relacionados con la variable respuesta. Una forma de evitar problemas de este tipo es, siempre que se excluyan predictores de un modelo, comprobar el impacto que tiene en su capacidad predictiva.
Incremento de la pureza de nodos
Cuantifica el incremento total en la pureza de los nodos debido a divisiones en las que participa el predictor (promedio de todos los árboles). La forma de calcularlo es la siguiente: en cada división de los árboles, se registra el descenso conseguido en la medida empleada como criterio de división (índice Gini, mse entropía, ...). Para cada uno de los predictores, se calcula el descenso medio conseguido en el conjunto de árboles que forman el ensemble. Cuanto mayor sea este valor medio, mayor la contribución del predictor en el modelo.
Ejemplo regresión¶
El set de datos Boston contiene precios de viviendas de la ciudad de Boston, así como información socio-económica del barrio en el que se encuentran. Se pretende ajustar un modelo de regresión que permita predecir el precio medio de una vivienda (MEDV) en función de las variables disponibles.
Librerías¶
Las librerías utilizadas en este ejemplo son:
# Tratamiento de datos
# ==============================================================================
import numpy as np
import pandas as pd
# Gráficos
# ==============================================================================
import matplotlib.pyplot as plt
plt.style.use('fivethirtyeight')
plt.rcParams['lines.linewidth'] = 1.5
plt.rcParams['font.size'] = 8
# Preprocesado y modelado
# ==============================================================================
import sklearn
from sklearn.ensemble import RandomForestRegressor
from sklearn.metrics import root_mean_squared_error
from sklearn.model_selection import cross_val_score
from sklearn.model_selection import train_test_split
from sklearn.model_selection import RepeatedKFold
from sklearn.model_selection import GridSearchCV
from sklearn.model_selection import ParameterGrid
from sklearn.model_selection import cross_val_score
from sklearn.inspection import permutation_importance
from joblib import Parallel, delayed, cpu_count
import optuna
# Configuración warnings
# ==============================================================================
import warnings
optuna.logging.set_verbosity(optuna.logging.WARNING)
print(f"Versión de scikit-learn: {sklearn.__version__}")
Versión de scikit-learn: 1.7.1
Datos¶
El set de datos Boston contiene precios de viviendas de la ciudad de Boston, así como información socio-económica del barrio en el que se encuentran. Se pretende ajustar un modelo de regresión que permita predecir el precio medio de una vivienda (MEDV) en función de las variables disponibles.
Número de instancias: 506
Número de atributos: 13 predictivos numéricos/categoriales.
Información de los atributos (por orden):
- CRIM: tasa de delincuencia per cápita por ciudad.
- ZN: proporción de suelo residencial dividido para segmentos de más de 25.000 pies cuadrados.
- INDUS: proporción de superficie comerciales no minoristas por ciudad.
- CHAS: variable binaria sobre el río Charles (= 1 si la zona linda con el río; 0 en caso contrario).
- NOX: concentración de óxidos nítricos (partes por 10 millones).
- RM: número medio de habitaciones por vivienda.
- EDAD: proporción de viviendas ocupadas construidas antes de 1940.
- DIS: distancia ponderada a cinco centros de empleo en la ciudad de Boston.
- RAD: índice de accesibilidad a las autopistas radiales.
- TAX: impuesto sobre bienes inmuebles de valor íntegro por cada 10.000$.
- PTRATIO: ratio alumnos-profesor por ciudad.
- LSTAT: % de la población considerado de clase baja.
- MEDV: valor medio de las viviendas ocupadas por sus propietarios en miles de $.
Valores ausentes: Ninguno
# Descarga de datos
# ==============================================================================
url = (
'https://raw.githubusercontent.com/JoaquinAmatRodrigo/Estadistica-machine-learning-python/'
'master/data/Boston.csv'
)
datos = pd.read_csv(url, sep=',')
display(datos.head(3))
display(datos.info())
| CRIM | ZN | INDUS | CHAS | NOX | RM | AGE | DIS | RAD | TAX | PTRATIO | LSTAT | MEDV | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0.00632 | 18.0 | 2.31 | 0 | 0.538 | 6.575 | 65.2 | 4.0900 | 1 | 296 | 15.3 | 4.98 | 24.0 |
| 1 | 0.02731 | 0.0 | 7.07 | 0 | 0.469 | 6.421 | 78.9 | 4.9671 | 2 | 242 | 17.8 | 9.14 | 21.6 |
| 2 | 0.02729 | 0.0 | 7.07 | 0 | 0.469 | 7.185 | 61.1 | 4.9671 | 2 | 242 | 17.8 | 4.03 | 34.7 |
<class 'pandas.core.frame.DataFrame'> RangeIndex: 506 entries, 0 to 505 Data columns (total 13 columns): # Column Non-Null Count Dtype --- ------ -------------- ----- 0 CRIM 506 non-null float64 1 ZN 506 non-null float64 2 INDUS 506 non-null float64 3 CHAS 506 non-null int64 4 NOX 506 non-null float64 5 RM 506 non-null float64 6 AGE 506 non-null float64 7 DIS 506 non-null float64 8 RAD 506 non-null int64 9 TAX 506 non-null int64 10 PTRATIO 506 non-null float64 11 LSTAT 506 non-null float64 12 MEDV 506 non-null float64 dtypes: float64(10), int64(3) memory usage: 51.5 KB
None
Ajuste del modelo¶
Se ajusta un modelo empleando como variable respuesta MEDV y como predictores todas las otras variables disponibles.
La clase RandomForestRegressor del módulo sklearn.ensemble permite entrenar modelos random forest para problemas de regresión. Los parámetros e hiperparámetros empleados por defecto son:
n_estimators=100criterion='squared_error'max_depth=Nonemin_samples_split=2min_samples_leaf=1min_weight_fraction_leaf=0.0max_features=1.0max_leaf_nodes=Nonemin_impurity_decrease=0.0bootstrap=Trueoob_score=Falsen_jobs=Nonerandom_state=Noneverbose=0warm_start=Falseccp_alpha=0.0max_samples=None
De entre todos ellos, destacan aquellos que detienen el crecimiento de los árboles, los que controlan el número de árboles y predictores incluidos, y los que gestionan la paralelización:
n_estimators; número de árboles incluidos en el modelo.max_depth: profundidad máxima que pueden alcanzar los árboles.min_samples_split: número mínimo de observaciones que debe de tener un nodo para que pueda dividirse. Si es un valor decimal se interpreta como fracción del total de observaciones de entrenamientoceil(min_samples_split * n_samples).min_samples_leaf: número mínimo de observaciones que debe de tener cada uno de los nodos hijos para que se produzca la división. Si es un valor decimal se interpreta como fracción del total de observaciones de entrenamientoceil(min_samples_split * n_samples).max_leaf_nodes: número máximo de nodos terminales que pueden tener los árboles.max_features: número de predictores considerados a en cada división. Puede ser:- Un valor entero
- Una fracción del total de predictores..
- “sqrt”, raiz cuadrada del número total de predictores.
- “log2”, log2 del número total de predictores.
- None, utiliza todos los predictores.
oob_score: Si se calcula o no el out-of-bag R^2. Por defecto esFalseya que aumenta el tiempo de entrenamiento.n_jobs: número de cores empleados para el entrenamiento. En random forest los árboles se ajustan de forma independiente, por lo la paralelización reduce notablemente el tiempo de entrenamiento. Con-1se utilizan todos los cores disponibles.random_state: semilla para que los resultados sean reproducibles. Tiene que ser un valor entero.
Como en todo estudio predictivo, no solo es importante ajustar el modelo, sino también cuantificar su capacidad para predecir nuevas observaciones. Para poder hacer esta evaluación, se dividen los datos en dos grupos, uno de entrenamiento y otro de test.
# División de los datos en entrenamiento y test
# ==============================================================================
X_train, X_test, y_train, y_test = train_test_split(
datos.drop(columns="MEDV"),
datos['MEDV'],
test_size = 0.25,
random_state = 123
)
print(f"Tamaño conjunto entrenamiento: {X_train.shape[0]}")
print(f"Tamaño conjunto test: {X_test.shape[0]}")
# Creación del modelo
# ==============================================================================
modelo = RandomForestRegressor(
n_estimators = 10,
criterion = 'squared_error',
max_depth = None,
max_features = 1,
oob_score = False,
n_jobs = -1,
random_state = 123
)
# Entrenamiento del modelo
# ==============================================================================
modelo.fit(X_train, y_train)
Tamaño conjunto entrenamiento: 379 Tamaño conjunto test: 127
RandomForestRegressor(max_features=1, n_estimators=10, n_jobs=-1,
random_state=123)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Parameters
| n_estimators | 10 | |
| criterion | 'squared_error' | |
| max_depth | None | |
| min_samples_split | 2 | |
| min_samples_leaf | 1 | |
| min_weight_fraction_leaf | 0.0 | |
| max_features | 1 | |
| max_leaf_nodes | None | |
| min_impurity_decrease | 0.0 | |
| bootstrap | True | |
| oob_score | False | |
| n_jobs | -1 | |
| random_state | 123 | |
| verbose | 0 | |
| warm_start | False | |
| ccp_alpha | 0.0 | |
| max_samples | None | |
| monotonic_cst | None |
Predicción y evaluación del modelo¶
Una vez entrenado el modelo, se evalúa la capacidad predictiva empleando el conjunto de test.
# Error de test del modelo inicial
# ==============================================================================
predicciones = modelo.predict(X=X_test)
rmse = root_mean_squared_error(y_true=y_test, y_pred=predicciones)
print(f"El error (rmse) de test es: {rmse}")
El error (rmse) de test es: 4.17800427058654
Optimización de hiperparámetros¶
El modelo inicial se ha entrenado utilizando 10 árboles (n_estimators=10) y manteniendo el resto de hiperparámetros con su valor por defecto. Al ser hiperparámetros, no se puede saber de antemano cuál es el valor más adecuado, la forma de identificarlos es mediante el uso de estrategias de validación, por ejemplo validación cruzada.
Los modelos Random Forest tienen la ventaja de disponer del Out-of-Bag error, lo que permite obtener una estimación del error de test sin recurrir a la validación cruzada, que es computacionalmente costosa. En la implementación de RandomForestRegressor, la métrica devuelta como oob_score es el $R^2$, si se desea otra, se tiene que recurrir al método oob_decision_function_() para obtener las predicciones y con ellas calcular la métrica de interés. Para una explicación más detallada consultar: Grid search de modelos Random Forest con out-of-bag error y early stopping.
Cabe tener en cuenta que, cuando se busca el valor óptimo de un hiperparámetro con dos métricas distintas, el resultado obtenido raramente es el mismo. Lo importante es que ambas métricas identifiquen las mismas regiones de interés.
Número de árboles¶
En Random Forest, el número de árboles no es un hiperparámetro crítico en cuanto que, añadir árboles, solo puede hacer que mejorar el resultado. En Random Forest no se produce overfitting por exceso de árboles. Sin embargo, añadir árboles una vez que la mejora se estabiliza es una perdida te recursos computacionales.
# Validación empleando el Out-of-Bag error
# ==============================================================================
warnings.filterwarnings('ignore')
train_scores = []
oob_scores = []
# Valores evaluados
estimator_range = range(1, 150, 5)
# Bucle para entrenar un modelo con cada valor de n_estimators y extraer su error
# de entrenamiento y de Out-of-Bag.
for n_estimators in estimator_range:
modelo = RandomForestRegressor(
n_estimators = n_estimators,
criterion = 'squared_error',
max_depth = None,
max_features = 1,
oob_score = True,
n_jobs = -1,
random_state = 123
)
modelo.fit(X_train, y_train)
train_scores.append(modelo.score(X_train, y_train))
oob_scores.append(modelo.oob_score_)
# Gráfico con la evolución de los errores
fig, ax = plt.subplots(figsize=(5, 3))
ax.plot(estimator_range, train_scores, label="train scores")
ax.plot(estimator_range, oob_scores, label="out-of-bag scores")
ax.plot(estimator_range[np.argmax(oob_scores)], max(oob_scores),
marker='o', color = "red", label="max score")
ax.set_ylabel("R^2")
ax.set_xlabel("n_estimators")
ax.set_title("Evolución del out-of-bag-error vs número árboles")
plt.legend();
print(f"Valor óptimo de n_estimators: {estimator_range[np.argmax(oob_scores)]}")
warnings.filterwarnings('default')
Valor óptimo de n_estimators: 66

# Validación empleando k-cross-validation y neg_root_mean_squared_error
# ==============================================================================
train_scores = []
cv_scores = []
# Valores evaluados
estimator_range = range(1, 150, 5)
# Bucle para entrenar un modelo con cada valor de n_estimators y extraer su error
# de entrenamiento y de k-cross-validation.
for n_estimators in estimator_range:
modelo = RandomForestRegressor(
n_estimators = n_estimators,
criterion = 'squared_error',
max_depth = None,
max_features = 1,
oob_score = False,
n_jobs = -1,
random_state = 123
)
# Error de entrenamiento
modelo.fit(X_train, y_train)
predicciones = modelo.predict(X=X_train)
rmse = root_mean_squared_error(
y_true = y_train,
y_pred = predicciones,
)
train_scores.append(rmse)
# Error de validación cruzada
scores = cross_val_score(
estimator = modelo,
X = X_train,
y = y_train,
scoring = 'neg_root_mean_squared_error',
cv = 5
)
# Se agregan los scores de cross_val_score() y se pasa a positivo
cv_scores.append(-1*scores.mean())
# Gráfico con la evolución de los errores
fig, ax = plt.subplots(figsize=(5, 3))
ax.plot(estimator_range, train_scores, label="train scores")
ax.plot(estimator_range, cv_scores, label="cv scores")
ax.plot(estimator_range[np.argmin(cv_scores)], min(cv_scores),
marker='o', color = "red", label="min score")
ax.set_ylabel("root_mean_squared_error")
ax.set_xlabel("n_estimators")
ax.set_title("Evolución del cv-error vs número árboles")
plt.legend();
print(f"Valor óptimo de n_estimators: {estimator_range[np.argmin(cv_scores)]}")
Valor óptimo de n_estimators: 96

Si bien el valor óptimo de las métricas se alcanza con 66 y 96 árboles, las curvas indican que, a partir de 20 árboles, el error de validación del modelo se estabiliza.
Max features¶
El valor de máx_features es uno de los hiperparámetros más importantes de random forest, ya que es el que permite controlar cuánto se decorrelacionan los árboles entre sí.
# Validación empleando el Out-of-Bag error
# ==============================================================================
train_scores = []
oob_scores = []
# Valores evaluados
max_features_range = range(1, X_train.shape[1] + 1, 1)
# Bucle para entrenar un modelo con cada valor de max_features y extraer su error
# de entrenamiento y de Out-of-Bag.
for max_features in max_features_range:
modelo = RandomForestRegressor(
n_estimators = 100,
criterion = 'squared_error',
max_depth = None,
max_features = max_features,
oob_score = True,
n_jobs = -1,
random_state = 123
)
modelo.fit(X_train, y_train)
train_scores.append(modelo.score(X_train, y_train))
oob_scores.append(modelo.oob_score_)
# Gráfico con la evolución de los errores
fig, ax = plt.subplots(figsize=(5, 3))
ax.plot(max_features_range, train_scores, label="train scores")
ax.plot(max_features_range, oob_scores, label="out-of-bag scores")
ax.plot(max_features_range[np.argmax(oob_scores)], max(oob_scores),
marker='o', color = "red")
ax.set_ylabel("R^2")
ax.set_xlabel("max_features")
ax.set_title("Evolución del out-of-bag-error vs número de predictores")
plt.legend();
print(f"Valor óptimo de max_features: {max_features_range[np.argmax(oob_scores)]}")
Valor óptimo de max_features: 4

# Validación empleando k-cross-validation y neg_root_mean_squared_error
# ==============================================================================
train_scores = []
cv_scores = []
# Valores evaluados
max_features_range = range(1, X_train.shape[1] + 1, 1)
# Bucle para entrenar un modelo con cada valor de max_features y extraer su error
# de entrenamiento y de k-cross-validation.
for max_features in max_features_range:
modelo = RandomForestRegressor(
n_estimators = 100,
criterion = 'squared_error',
max_depth = None,
max_features = max_features,
oob_score = True,
n_jobs = -1,
random_state = 123
)
# Error de train
modelo.fit(X_train, y_train)
predicciones = modelo.predict(X = X_train)
rmse = root_mean_squared_error(
y_true = y_train,
y_pred = predicciones,
)
train_scores.append(rmse)
# Error de validación cruzada
scores = cross_val_score(
estimator = modelo,
X = X_train,
y = y_train,
scoring = 'neg_root_mean_squared_error',
cv = 5
)
# Se agregan los scores de cross_val_score() y se pasa a positivo
cv_scores.append(-1*scores.mean())
# Gráfico con la evolución de los errores
fig, ax = plt.subplots(figsize=(5, 3))
ax.plot(max_features_range, train_scores, label="train scores")
ax.plot(max_features_range, cv_scores, label="cv scores")
ax.plot(max_features_range[np.argmin(cv_scores)], min(cv_scores),
marker='o', color = "red", label="min score")
ax.set_ylabel("root_mean_squared_error")
ax.set_xlabel("max_features")
ax.set_title("Evolución del cv-error vs número de predictores")
plt.legend();
print(f"Valor óptimo de max_features: {max_features_range[np.argmin(cv_scores)]}")
Valor óptimo de max_features: 6

Acorde a las dos métricas utilizadas, el valor óptimo de max_features está entre 4 y 6.
Grid search¶
Aunque el análisis individual de los hiperparámetros es útil para entender su impacto en el modelo e identificar rangos de interés, la búsqueda final no debe hacerse de forma secuencial, ya que cada hiperparámetro interacciona con los demás. Es preferible recurrir a grid search, random search o bayesian search para analizar varias combinaciones de hiperparámetros. Puede encontrarse más información sobre las estrategias de búsqueda en Machine learning con Python y Scikit-learn.
Out-of-bag error¶
Más detalles en: Grid search de modelos Random Forest con out-of-bag error y early stopping.
# Grid de hiperparámetros evaluados
# ==============================================================================
param_grid = ParameterGrid(
{'n_estimators': [150],
'max_features': [5, 7, 9],
'max_depth' : [None, 3, 10, 20]
}
)
# Loop para ajustar un modelo con cada combinación de hiperparámetros
# ==============================================================================
resultados = {'params': [], 'oob_r2': []}
for params in param_grid:
modelo = RandomForestRegressor(
oob_score = True,
n_jobs = -1,
random_state = 123,
** params
)
modelo.fit(X_train, y_train)
resultados['params'].append(params)
resultados['oob_r2'].append(modelo.oob_score_)
print(f"Modelo: {params} ✓")
# Resultados
# ==============================================================================
resultados = pd.DataFrame(resultados)
resultados = pd.concat([resultados, resultados['params'].apply(pd.Series)], axis=1)
resultados = resultados.drop(columns = 'params')
resultados = resultados.sort_values('oob_r2', ascending=False)
resultados.head(4)
Modelo: {'max_depth': None, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'max_depth': None, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'max_depth': None, 'max_features': 9, 'n_estimators': 150} ✓
Modelo: {'max_depth': 3, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'max_depth': 3, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'max_depth': 3, 'max_features': 9, 'n_estimators': 150} ✓
Modelo: {'max_depth': 10, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'max_depth': 10, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'max_depth': 10, 'max_features': 9, 'n_estimators': 150} ✓
Modelo: {'max_depth': 20, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'max_depth': 20, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'max_depth': 20, 'max_features': 9, 'n_estimators': 150} ✓
| oob_r2 | max_depth | max_features | n_estimators | |
|---|---|---|---|---|
| 6 | 0.882461 | 10.0 | 5.0 | 150.0 |
| 0 | 0.875289 | NaN | 5.0 | 150.0 |
| 9 | 0.874590 | 20.0 | 5.0 | 150.0 |
| 10 | 0.874318 | 20.0 | 7.0 | 150.0 |
Este proceso de búsqueda puede paralelizarse para aprovechar todos cores del ordenador.
# Grid de hiperparámetros evaluados
# ==============================================================================
param_grid = ParameterGrid(
{'n_estimators': [150],
'max_features': [5, 7, 9],
'max_depth' : [None, 3, 10, 20]
}
)
# Loop paralelizado para ajustar un modelo con cada combinación de hiperparámetros
# ==============================================================================
def eval_oob_error(X, y, params, verbose=True):
"""
Función para entrenar un modelo utilizando unos parámetros determinados
y que devuelve el out-of-bag error
"""
modelo = RandomForestRegressor(
oob_score = True,
n_jobs = -1,
random_state = 123,
** params
)
modelo.fit(X, y)
if verbose:
print(f"Modelo: {params} ✓")
return{'params': params, 'oob_r2': modelo.oob_score_}
resultados = Parallel(n_jobs=cpu_count()-1)(
delayed(eval_oob_error)(X_train, y_train, params)
for params in param_grid
)
# Resultados
# ==============================================================================
resultados = pd.DataFrame(resultados)
resultados = pd.concat([resultados, resultados['params'].apply(pd.Series)], axis=1)
resultados = resultados.drop(columns = 'params')
resultados = resultados.sort_values('oob_r2', ascending=False)
resultados.head(4)
Modelo: {'max_depth': None, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'max_depth': 3, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'max_depth': 3, 'max_features': 9, 'n_estimators': 150} ✓
Modelo: {'max_depth': None, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'max_depth': None, 'max_features': 9, 'n_estimators': 150} ✓
Modelo: {'max_depth': 10, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'max_depth': 3, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'max_depth': 10, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'max_depth': 10, 'max_features': 9, 'n_estimators': 150} ✓
Modelo: {'max_depth': 20, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'max_depth': 20, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'max_depth': 20, 'max_features': 9, 'n_estimators': 150} ✓
| oob_r2 | max_depth | max_features | n_estimators | |
|---|---|---|---|---|
| 6 | 0.882461 | 10.0 | 5.0 | 150.0 |
| 0 | 0.875289 | NaN | 5.0 | 150.0 |
| 9 | 0.874590 | 20.0 | 5.0 | 150.0 |
| 10 | 0.874318 | 20.0 | 7.0 | 150.0 |
# Mejores hiperparámetros por out-of-bag error
# ==============================================================================
print("--------------------------------------------")
print("Mejores hiperparámetros encontrados (oob-r2)")
print("--------------------------------------------")
print(resultados.iloc[0,0:])
-------------------------------------------- Mejores hiperparámetros encontrados (oob-r2) -------------------------------------------- oob_r2 0.882461 max_depth 10.000000 max_features 5.000000 n_estimators 150.000000 Name: 6, dtype: float64
Validación cruzada¶
⚠ Warning
La busqueda de hiperparámetros debe hacerse utilizando datos que el modelo no ha visto. Si se emplean los mismos datos para ajustar el modelo y para evaluarlo, se corre el riesgo de sobreajustar el modelo a los datos de entrenamiento. Para evitar este problema se puede recurrir a dos estrategias:
Dividir el conjunto de datos en tres grupos: entrenamiento, validación y test. El conjunto de entrenamiento se emplea para ajustar el modelo, el de validación para seleccionar el valor óptimo de los hiperparámetros y el de test para evaluar la capacidad predictiva del modelo.
Utilizar validación cruzada con los datos de entrenamiento. En este caso, el conjunto de entrenamiento se divide en k grupos y se ajusta el modelo k veces, cada vez con un grupo distinto como conjunto de validación. La métrica final es el promedio de los valores obtenidos en cada iteración. Esta estrategia es computacionalmente más costosa, ya que requiere entrenar el modelo k veces, pero evita tener que crear una partición adicional de los datos cuando no se dispone de muchos.
# Grid de hiperparámetros evaluados
# ==============================================================================
param_grid = {'n_estimators': [150],
'max_features': [5, 7, 9],
'max_depth' : [None, 3, 10, 20]
}
# Búsqueda por grid search con validación cruzada
# ==============================================================================
grid = GridSearchCV(
estimator = RandomForestRegressor(random_state = 123),
param_grid = param_grid,
scoring = 'neg_root_mean_squared_error',
n_jobs = cpu_count() - 1,
cv = RepeatedKFold(n_splits=5, n_repeats=3, random_state=123),
refit = True,
verbose = 0,
return_train_score = True
)
grid.fit(X=X_train, y=y_train)
# Resultados
# ==============================================================================
resultados = pd.DataFrame(grid.cv_results_)
resultados.filter(regex = '(param.*|mean_t|std_t)') \
.drop(columns = 'params') \
.sort_values('mean_test_score', ascending = False) \
.head(4)
| param_max_depth | param_max_features | param_n_estimators | mean_test_score | std_test_score | mean_train_score | std_train_score | |
|---|---|---|---|---|---|---|---|
| 6 | 10 | 5 | 150 | -3.320295 | 0.701794 | -1.323499 | 0.061678 |
| 7 | 10 | 7 | 150 | -3.333357 | 0.696114 | -1.327214 | 0.066324 |
| 0 | None | 5 | 150 | -3.334407 | 0.715278 | -1.261911 | 0.058096 |
| 9 | 20 | 5 | 150 | -3.336755 | 0.716904 | -1.261439 | 0.057603 |
# Mejores hiperparámetros encontrados mediante validación cruzada
# ==============================================================================
print("----------------------------------------")
print("Mejores hiperparámetros encontrados (cv)")
print("----------------------------------------")
print(grid.best_params_, ":", grid.best_score_, grid.scoring)
----------------------------------------
Mejores hiperparámetros encontrados (cv)
----------------------------------------
{'max_depth': 10, 'max_features': 5, 'n_estimators': 150} : -3.320294830700744 neg_root_mean_squared_error
Una vez identificados los mejores hiperparámetros, se reentrena el modelo indicando los valores óptimos en sus argumentos. Si en el GridSearchCV() se indica refit=True, este reentrenamiento se hace automáticamente y
el modelo resultante se encuentra almacenado en .best_estimator_.
# Error de test del modelo final
# ==============================================================================
modelo_final = grid.best_estimator_
predicciones = modelo_final.predict(X=X_test)
rmse = root_mean_squared_error(
y_true = y_test,
y_pred = predicciones,
)
print(f"El error (rmse) de test es: {rmse}")
El error (rmse) de test es: 3.550875871674505
Tras optimizar los hiperparámetros, se consigue reducir el error rmse del modelo de 4.35 a 3.55. Las predicciones del modelo final se alejan en promedio 3.55 unidades (3550 dólares) del valor real.
Bayesian search¶
La búsqueda grid search y random search pueden generar buenos resultados, sobre todo cuando se reduce el rango de exploración. Sin embargo, ninguna de ellas tiene en cuenta los resultados obtenidos hasta el momento, lo que les impide centrar la búsqueda en las regiones de mayor interés y evitar las innecesarias.
Una alternativa es utilizar métodos de optimización bayesiana para buscar hiperparámetros. En términos generales, la optimización bayesiana de hiperparámetros consiste en crear un modelo probabilístico en el que la función objetivo es la métrica de validación del modelo (RMSE, AUC, precisión...). Con esta estrategia, la búsqueda se redirige en cada iteración a las regiones de mayor interés. El objetivo final es reducir el número de combinaciones de hiperparámetros con las que se evalúa el modelo, eligiendo sólo los mejores candidatos. Este enfoque es especialmente ventajoso cuando el espacio de búsqueda es muy grande o la evaluación del modelo es muy lenta.
# Búsqueda bayesiana de hiperparámetros con optuna
# ==============================================================================
def objective(trial):
params = {
'n_estimators': trial.suggest_int('n_estimators', 100, 1000, step=100),
'max_depth': trial.suggest_int('max_depth', 3, 30),
'min_samples_split': trial.suggest_int('min_samples_split', 2, 100),
'min_samples_leaf': trial.suggest_int('min_samples_leaf', 1, 100),
'max_features': trial.suggest_float('max_features', 0.2, 1.0),
'ccp_alpha': trial.suggest_float('ccp_alpha', 0.0, 1),
# Fixed parameters
'n_jobs': -1,
'random_state': 4576688,
}
modelo = RandomForestRegressor(**params)
cross_val_scores = cross_val_score(
estimator = modelo,
X = X_train,
y = y_train,
cv = RepeatedKFold(n_splits=5, n_repeats=3, random_state=123),
scoring = 'neg_root_mean_squared_error',
n_jobs=-1
)
score = np.mean(cross_val_scores)
return score
study = optuna.create_study(direction='maximize') # Se maximiza por que el score es negativo
study.optimize(objective, n_trials=30, show_progress_bar=True, timeout=60*5)
print('Mejores hyperparameters:', study.best_params)
print('Mejor score:', study.best_value)
0%| | 0/30 [00:00<?, ?it/s]
Mejores hyperparameters: {'n_estimators': 200, 'max_depth': 19, 'min_samples_split': 23, 'min_samples_leaf': 10, 'max_features': 0.5010767895372719, 'ccp_alpha': 0.592918283831454}
Mejor score: -4.077406292401763
# Error de test del modelo final
# ==============================================================================
modelo_final = RandomForestRegressor(**study.best_params)
modelo_final.fit(X_train, y_train)
predicciones = modelo_final.predict(X=X_test)
rmse = root_mean_squared_error(y_true=y_test, y_pred=predicciones)
print(f"El error (rmse) de test es: {rmse}")
El error (rmse) de test es: 4.458933398642248
Importancia de predictores¶
Pureza de nodos¶
# Importancia de los predictores basada en la reducción de la impureza
# ==============================================================================
importancia_predictores = pd.DataFrame({
'predictor': datos.drop(columns = "MEDV").columns,
'importancia': modelo_final.feature_importances_
})
print("Importancia de los predictores en el modelo")
print("-------------------------------------------")
importancia_predictores.sort_values('importancia', ascending=False)
Importancia de los predictores en el modelo -------------------------------------------
| predictor | importancia | |
|---|---|---|
| 5 | RM | 0.399866 |
| 11 | LSTAT | 0.378344 |
| 4 | NOX | 0.061980 |
| 10 | PTRATIO | 0.038606 |
| 0 | CRIM | 0.037579 |
| 2 | INDUS | 0.036500 |
| 7 | DIS | 0.026432 |
| 9 | TAX | 0.011672 |
| 6 | AGE | 0.006530 |
| 1 | ZN | 0.001505 |
| 8 | RAD | 0.000986 |
| 3 | CHAS | 0.000000 |
Permutación¶
# Importancia de los predictores basada en permutación
# ==============================================================================
importancia = permutation_importance(
estimator = modelo_final,
X = X_train,
y = y_train,
n_repeats = 5,
scoring = 'neg_root_mean_squared_error',
n_jobs = cpu_count() - 1,
random_state = 123
)
# Se almacenan los resultados (media y desviación) en un dataframe
df_importancia = pd.DataFrame(
{k: importancia[k] for k in ['importances_mean', 'importances_std']}
)
df_importancia['feature'] = X_train.columns
df_importancia.sort_values('importances_mean', ascending=False)
| importances_mean | importances_std | feature | |
|---|---|---|---|
| 11 | 3.463219 | 0.156903 | LSTAT |
| 5 | 3.195968 | 0.077292 | RM |
| 4 | 0.391688 | 0.049401 | NOX |
| 0 | 0.240019 | 0.030626 | CRIM |
| 10 | 0.228540 | 0.028866 | PTRATIO |
| 7 | 0.160030 | 0.021386 | DIS |
| 2 | 0.110869 | 0.013235 | INDUS |
| 9 | 0.068014 | 0.006425 | TAX |
| 6 | 0.040218 | 0.005447 | AGE |
| 8 | 0.015989 | 0.002731 | RAD |
| 1 | 0.004424 | 0.001648 | ZN |
| 3 | 0.000000 | 0.000000 | CHAS |
# Gráfico de la importancia de cada predictor
# ==============================================================================
fig, ax = plt.subplots(figsize=(3.5, 4))
df_importancia = df_importancia.sort_values('importances_mean', ascending=True)
ax.barh(
df_importancia['feature'],
df_importancia['importances_mean'],
xerr=df_importancia['importances_std'],
align='center',
alpha=0
)
ax.plot(
df_importancia['importances_mean'],
df_importancia['feature'],
marker="D",
linestyle="",
alpha=0.8,
color="r"
)
ax.set_title('Importancia de los predictores (train)')
ax.set_xlabel('Incremento del error tras la permutación');

Ambas estrategias identifican LSTAT y RM como los predictores más influyentes, acorde a los datos de entrenamiento.
Ejemplo clasificación¶
Librerías¶
Las librerías utilizadas en este documento son:
# Tratamiento de datos
# ==============================================================================
import numpy as np
import pandas as pd
import statsmodels.api as sm
# Gráficos
# ==============================================================================
import matplotlib.pyplot as plt
plt.style.use('fivethirtyeight')
plt.rcParams['lines.linewidth'] = 1.5
plt.rcParams['font.size'] = 8
# Preprocesado y modelado
# ==============================================================================
from sklearn.ensemble import RandomForestClassifier
from sklearn.metrics import accuracy_score
from sklearn.metrics import confusion_matrix
from sklearn.metrics import ConfusionMatrixDisplay
from sklearn.metrics import classification_report
from sklearn.compose import ColumnTransformer
from sklearn.preprocessing import OneHotEncoder
from sklearn.model_selection import cross_val_score
from sklearn.model_selection import train_test_split
from sklearn.model_selection import RepeatedKFold
from sklearn.model_selection import GridSearchCV
from sklearn.model_selection import ParameterGrid
from sklearn.inspection import permutation_importance
from joblib import Parallel, delayed
import optuna
# Configuración warnings
# ==============================================================================
import warnings
Datos¶
El set de datos Carseats, original del paquete de R ISLR y accesible en Python a través de statsmodels.datasets.get_rdataset, contiene información sobre la venta de sillas infantiles en 400 tiendas distintas. Para cada una de las 400 tiendas se han registrado 11 variables. Se pretende generar un modelo de clasificación que permita predecir si una tienda tiene ventas altas (Sales > 8) o bajas (Sales <= 8) en función de todas las variables disponibles.
✎ Nota
Listado de todos los set de datos disponibles en Rdatasets.# Lectura de datos
# ==============================================================================
carseats = sm.datasets.get_rdataset("Carseats", "ISLR")
datos = carseats.data
print(carseats.__doc__)
.. container::
.. container::
======== ===============
Carseats R Documentation
======== ===============
.. rubric:: Sales of Child Car Seats
:name: sales-of-child-car-seats
.. rubric:: Description
:name: description
A simulated data set containing sales of child car seats at 400
different stores.
.. rubric:: Usage
:name: usage
.. code:: R
Carseats
.. rubric:: Format
:name: format
A data frame with 400 observations on the following 11 variables.
``Sales``
Unit sales (in thousands) at each location
``CompPrice``
Price charged by competitor at each location
``Income``
Community income level (in thousands of dollars)
``Advertising``
Local advertising budget for company at each location (in
thousands of dollars)
``Population``
Population size in region (in thousands)
``Price``
Price company charges for car seats at each site
``ShelveLoc``
A factor with levels ``Bad``, ``Good`` and ``Medium``
indicating the quality of the shelving location for the car
seats at each site
``Age``
Average age of the local population
``Education``
Education level at each location
``Urban``
A factor with levels ``No`` and ``Yes`` to indicate whether the
store is in an urban or rural location
``US``
A factor with levels ``No`` and ``Yes`` to indicate whether the
store is in the US or not
.. rubric:: Source
:name: source
Simulated data
.. rubric:: References
:name: references
James, G., Witten, D., Hastie, T., and Tibshirani, R. (2013) *An
Introduction to Statistical Learning with applications in R*,
https://www.statlearning.com, Springer-Verlag, New York
.. rubric:: Examples
:name: examples
.. code:: R
summary(Carseats)
lm.fit=lm(Sales~Advertising+Price,data=Carseats)
datos.head(3)
| Sales | CompPrice | Income | Advertising | Population | Price | ShelveLoc | Age | Education | Urban | US | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 9.50 | 138 | 73 | 11 | 276 | 120 | Bad | 42 | 17 | Yes | Yes |
| 1 | 11.22 | 111 | 48 | 16 | 260 | 83 | Good | 65 | 10 | Yes | Yes |
| 2 | 10.06 | 113 | 35 | 10 | 269 | 80 | Medium | 59 | 12 | Yes | Yes |
Como Sales es una variable continua y el objetivo del estudio es clasificar las tiendas según si venden mucho o poco, se crea una nueva variable binaria (0, 1) llamada ventas_altas.
datos['ventas_altas'] = np.where(datos.Sales > 8, 0, 1)
# Una vez creada la nueva variable respuesta se descarta la original
datos = datos.drop(columns = 'Sales')
Ajuste y optimización de hiperparámetros¶
Se ajusta un árbol de clasificación empleando como variable respuesta ventas_altas y como predictores todas las variables disponibles. Se utilizan en primer lugar los hiperparámetros max_depth=5 y criterion='gini', el resto se dejan por defecto. Después, se aplica el proceso de pruning y se comparan los resultados frente al modelo inicial.
A diferencia del ejemplo anterior, en estos datos hay variables categóricas por lo que, antes de entrenar el modelo, es necesario aplicar one-hot-encoding. Puede encontrarse una descripción más detallada de este proceso en Machine learning con Python y Scikit-learn.
# División de los datos en train y test
# ==============================================================================
X_train, X_test, y_train, y_test = train_test_split(
datos.drop(columns = 'ventas_altas'),
datos['ventas_altas'],
random_state = 123
)
# One-hot-encoding de las variables categóricas
# ==============================================================================
# Se identifica el nobre de las columnas numéricas y categóricas
cat_cols = X_train.select_dtypes(include=['object', 'category']).columns.to_list()
numeric_cols = X_train.select_dtypes(include=['float64', 'int']).columns.to_list()
# Se aplica one-hot-encoding solo a las columnas categóricas
preprocessor = ColumnTransformer(
[('onehot', OneHotEncoder(handle_unknown='ignore', sparse_output=False, drop='if_binary'), cat_cols)],
remainder='passthrough',
verbose_feature_names_out=False
).set_output(transform="pandas")
# Una vez que se ha definido el objeto ColumnTransformer, con el método fit()
# se aprenden las transformaciones con los datos de entrenamiento y se aplican a
# los dos conjuntos con transform(). Ambas operaciones a la vez con fit_transform().
X_train_prep = preprocessor.fit_transform(X_train)
X_test_prep = preprocessor.transform(X_test)
X_train_prep.info()
<class 'pandas.core.frame.DataFrame'> Index: 300 entries, 170 to 365 Data columns (total 12 columns): # Column Non-Null Count Dtype --- ------ -------------- ----- 0 ShelveLoc_Bad 300 non-null float64 1 ShelveLoc_Good 300 non-null float64 2 ShelveLoc_Medium 300 non-null float64 3 Urban_Yes 300 non-null float64 4 US_Yes 300 non-null float64 5 CompPrice 300 non-null int64 6 Income 300 non-null int64 7 Advertising 300 non-null int64 8 Population 300 non-null int64 9 Price 300 non-null int64 10 Age 300 non-null int64 11 Education 300 non-null int64 dtypes: float64(5), int64(7) memory usage: 30.5 KB
Si bien RandomForestClassifier tiene valores por defecto para sus hiperparámetros, no se puede saber de antemano si estos son los más adecuados, la forma de identificarlos es mediante el uso de estrategias de validación, por ejemplo validación cruzada.
Los modelos Random Forest tienen la ventaja de disponer del Out-of-Bag error, lo que permite obtener una estimación del error de test sin recurrir a la validación cruzada, que es computacionalmente costosa. En la implementación de RandomForestClassifier, la métrica devuelta como oob_score es el accuracy, si se desea otra, se tiene que recurrir al método oob_decision_function_() para obtener las predicciones y con ellas calcular la métrica de interés. Para una explicación más detallada consultar: Grid search de modelos Random Forest con out-of-bag error y early stopping.
Cabe tener en cuenta que, cuando se busca el valor óptimo de un hiperparámetro con dos métricas distintas, el resultado obtenido raramente es el mismo. Lo importante es que ambas métricas identifiquen las mismas regiones de interés.
Aunque el análisis individual de los hiperparámetros es útil para entender su impacto en el modelo e identificar rangos de interés, la búsqueda final no debe hacerse de forma secuencial, ya que cada hiperparámetro interacciona con los demás. Es preferible recurrir a grid search o random search para analizar varias combinaciones de hiperparámetros. Puede encontrarse más información sobre las estrategias de búsqueda en Machine learning con Python y Scikit-learn.
Grid Search (out-of-bag score)¶
Más detalles en: Grid search de modelos Random Forest con out-of-bag error y early stopping.
# Grid de hiperparámetros evaluados
# ==============================================================================
param_grid = ParameterGrid(
{'n_estimators': [150],
'max_features': [5, 7, 9],
'max_depth' : [None, 3, 10, 20],
'criterion' : ['gini', 'entropy']
}
)
# Loop para ajustar un modelo con cada combinación de hiperparámetros
# ==============================================================================
resultados = {'params': [], 'oob_accuracy': []}
for params in param_grid:
modelo = RandomForestClassifier(
oob_score = True,
n_jobs = -1,
random_state = 123,
** params
)
modelo.fit(X_train_prep, y_train)
resultados['params'].append(params)
resultados['oob_accuracy'].append(modelo.oob_score_)
print(f"Modelo: {params} \u2713")
# Resultados
# ==============================================================================
resultados = pd.DataFrame(resultados)
resultados = pd.concat([resultados, resultados['params'].apply(pd.Series)], axis=1)
resultados = resultados.sort_values('oob_accuracy', ascending=False)
resultados = resultados.drop(columns='params')
resultados.head(4)
Modelo: {'criterion': 'gini', 'max_depth': None, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'criterion': 'gini', 'max_depth': None, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'criterion': 'gini', 'max_depth': None, 'max_features': 9, 'n_estimators': 150} ✓
Modelo: {'criterion': 'gini', 'max_depth': 3, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'criterion': 'gini', 'max_depth': 3, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'criterion': 'gini', 'max_depth': 3, 'max_features': 9, 'n_estimators': 150} ✓
Modelo: {'criterion': 'gini', 'max_depth': 10, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'criterion': 'gini', 'max_depth': 10, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'criterion': 'gini', 'max_depth': 10, 'max_features': 9, 'n_estimators': 150} ✓
Modelo: {'criterion': 'gini', 'max_depth': 20, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'criterion': 'gini', 'max_depth': 20, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'criterion': 'gini', 'max_depth': 20, 'max_features': 9, 'n_estimators': 150} ✓
Modelo: {'criterion': 'entropy', 'max_depth': None, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'criterion': 'entropy', 'max_depth': None, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'criterion': 'entropy', 'max_depth': None, 'max_features': 9, 'n_estimators': 150} ✓
Modelo: {'criterion': 'entropy', 'max_depth': 3, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'criterion': 'entropy', 'max_depth': 3, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'criterion': 'entropy', 'max_depth': 3, 'max_features': 9, 'n_estimators': 150} ✓
Modelo: {'criterion': 'entropy', 'max_depth': 10, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'criterion': 'entropy', 'max_depth': 10, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'criterion': 'entropy', 'max_depth': 10, 'max_features': 9, 'n_estimators': 150} ✓
Modelo: {'criterion': 'entropy', 'max_depth': 20, 'max_features': 5, 'n_estimators': 150} ✓
Modelo: {'criterion': 'entropy', 'max_depth': 20, 'max_features': 7, 'n_estimators': 150} ✓
Modelo: {'criterion': 'entropy', 'max_depth': 20, 'max_features': 9, 'n_estimators': 150} ✓
| oob_accuracy | criterion | max_depth | max_features | n_estimators | |
|---|---|---|---|---|---|
| 22 | 0.820000 | entropy | 20.0 | 7 | 150 |
| 13 | 0.820000 | entropy | NaN | 7 | 150 |
| 0 | 0.813333 | gini | NaN | 5 | 150 |
| 6 | 0.813333 | gini | 10.0 | 5 | 150 |
# Mejores hiperparámetros por out-of-bag error
# ==============================================================================
print("--------------------------------------------------")
print("Mejores hiperparámetros encontrados (oob-accuracy)")
print("--------------------------------------------------")
print(resultados.iloc[0,0:])
-------------------------------------------------- Mejores hiperparámetros encontrados (oob-accuracy) -------------------------------------------------- oob_accuracy 0.82 criterion entropy max_depth 20.0 max_features 7 n_estimators 150 Name: 22, dtype: object
Grid Search (validación cruzada)¶
# Grid de hiperparámetros evaluados
# ==============================================================================
param_grid = {
'n_estimators': [150],
'max_features': [5, 7, 9],
'max_depth' : [None, 3, 10, 20],
'criterion' : ['gini', 'entropy']
}
# Búsqueda por grid search con validación cruzada
# ==============================================================================
grid = GridSearchCV(
estimator = RandomForestClassifier(random_state = 123),
param_grid = param_grid,
scoring = 'accuracy',
n_jobs = cpu_count() - 1,
cv = RepeatedKFold(n_splits=5, n_repeats=3, random_state=123),
refit = True,
verbose = 0,
return_train_score = True
)
grid.fit(X=X_train_prep, y=y_train)
# Resultados
# ==============================================================================
resultados = pd.DataFrame(grid.cv_results_)
resultados.filter(regex='(param*|mean_t|std_t)') \
.drop(columns='params') \
.sort_values('mean_test_score', ascending = False) \
.head(4)
| param_criterion | param_max_depth | param_max_features | param_n_estimators | mean_test_score | std_test_score | mean_train_score | std_train_score | |
|---|---|---|---|---|---|---|---|---|
| 21 | entropy | 20 | 5 | 150 | 0.817778 | 0.028846 | 1.0 | 0.0 |
| 12 | entropy | None | 5 | 150 | 0.817778 | 0.028846 | 1.0 | 0.0 |
| 18 | entropy | 10 | 5 | 150 | 0.814444 | 0.030952 | 1.0 | 0.0 |
| 13 | entropy | None | 7 | 150 | 0.812222 | 0.027532 | 1.0 | 0.0 |
# Mejores hiperparámetros encontrados por validación cruzada
# ==============================================================================
print("--------------------------------------------")
print("Mejores hiperparámetros encontrados por (cv)")
print("--------------------------------------------")
print(grid.best_params_, ":", grid.best_score_, grid.scoring)
--------------------------------------------
Mejores hiperparámetros encontrados por (cv)
--------------------------------------------
{'criterion': 'entropy', 'max_depth': None, 'max_features': 5, 'n_estimators': 150} : 0.8177777777777779 accuracy
Una vez identificados los mejores hiperparámetros, se reentrena el modelo indicando los valores óptimos en sus argumentos. Si en el GridSearchCV() se indica refit=True, este reentrenamiento se hace automáticamente y
el modelo resultante se encuentra almacenado en .best_estimator_.
Predicción y evaluación¶
Por último, se evalúa la capacidad predictiva del modelo final empleando el conjunto de test.
# Modelo con los mejores hiperparámetros
# ==============================================================================
modelo_final = grid.best_estimator_
modelo_final
RandomForestClassifier(criterion='entropy', max_features=5, n_estimators=150,
random_state=123)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Parameters
| n_estimators | 150 | |
| criterion | 'entropy' | |
| max_depth | None | |
| min_samples_split | 2 | |
| min_samples_leaf | 1 | |
| min_weight_fraction_leaf | 0.0 | |
| max_features | 5 | |
| max_leaf_nodes | None | |
| min_impurity_decrease | 0.0 | |
| bootstrap | True | |
| oob_score | False | |
| n_jobs | None | |
| random_state | 123 | |
| verbose | 0 | |
| warm_start | False | |
| class_weight | None | |
| ccp_alpha | 0.0 | |
| max_samples | None | |
| monotonic_cst | None |
# Error de test del modelo final
# ==============================================================================
predicciones = modelo_final.predict(X=X_test_prep)
predicciones[:10]
array([0, 0, 0, 0, 0, 0, 1, 1, 1, 1])
mat_confusion = confusion_matrix(y_true=y_test, y_pred=predicciones)
accuracy = accuracy_score(y_true=y_test, y_pred=predicciones, normalize=True)
print("Matriz de confusión")
print("-------------------")
print(mat_confusion)
print("")
print(f"El accuracy de test es: {100 * accuracy} % \n")
fig, ax = plt.subplots(figsize=(3, 3))
ConfusionMatrixDisplay(mat_confusion).plot(ax=ax)
print(
classification_report(
y_true = y_test,
y_pred = predicciones
)
)
Matriz de confusión
-------------------
[[33 17]
[ 3 47]]
El accuracy de test es: 80.0 %
precision recall f1-score support
0 0.92 0.66 0.77 50
1 0.73 0.94 0.82 50
accuracy 0.80 100
macro avg 0.83 0.80 0.80 100
weighted avg 0.83 0.80 0.80 100

Tras optimizar los hiperparámetros, se consigue un porcentaje de acierto del 78%.
Predicción probabilística¶
La mayoría de implementaciones de Random Forest, entre ellas la de scikit-learn, permiten predecir probabilidades cuando se trata de problemas de clasificación. Es importante entender cómo se calculan estos valores para interpretarlos y utilizarlos correctamente.
En el ejemplo anterior, al aplicar .predict() se devuelve $1$ (ventas elevadas) o $0$ (ventas bajas) para cada observación de test. Sin embargo, no se dispone de ningún tipo de información sobre la seguridad con la que el modelo realiza esta asignación. Con .predict_proba(), en lugar de una clasificación, se obtiene la probabilidad con la que el modelo considera que cada observación puede pertenecer a cada una de las clases.
# Predicción de probabilidades
# ==============================================================================
predicciones = modelo_final.predict_proba(X=X_test_prep)
predicciones[:5, :]
array([[0.78666667, 0.21333333],
[0.72 , 0.28 ],
[0.86 , 0.14 ],
[0.72666667, 0.27333333],
[0.79333333, 0.20666667]])
El resultado de .predict_proba() es un array con una fila por observación y tantas columnas como clases tenga la variable respuesta. El valor de la primera columna se corresponde con la probabilidad, acorde al modelo, de que la observación pertenezca a la clase 0, y así sucesivamente. El valor de probabilidad mostrado para cada predicción se corresponde con la fracción de observaciones de cada clase en los nodos terminales a los que ha llegado la observación predicha en el conjunto de los árboles.
Por defecto, .predict() asigna cada nueva observación a la clase con mayor probabilidad (en caso de empate se asigna de forma aleatoria). Sin embargo, este no tiene por qué ser el comportamiento deseado en todos los casos.
# Clasificación empleando la clase de mayor probabilidad
# ==============================================================================
df_predicciones = pd.DataFrame(data=predicciones, columns=['0', '1'])
df_predicciones['clasificacion_default_0.5'] = np.where(df_predicciones['0'] > df_predicciones['1'], 0, 1)
df_predicciones.head(3)
| 0 | 1 | clasificacion_default_0.5 | |
|---|---|---|---|
| 0 | 0.786667 | 0.213333 | 0 |
| 1 | 0.720000 | 0.280000 | 0 |
| 2 | 0.860000 | 0.140000 | 0 |
Supóngase el siguiente escenario: la campaña de navidad se aproxima y los propietarios de la cadena quieren duplicar el stock de artículos en aquellas tiendas de las que se preve que tengan ventas elevadas. Como el transporte de este material hasta las tiendas supone un coste elevado, el director quiere limitar esta estrategia únicamente a tiendas para las que se tenga mucha seguridad de que van conseguir muchas ventas.
Si se dispone de las probabilidades, se puede establecer un punto de corte concreto, por ejemplo, considerando únicamente como clase $1$ (ventas altas) aquellas tiendas cuya predicción para esta clase sea superior al 0.9 (90%). De esta forma, la clasificación final se ajusta mejor a las necesidades del caso de uso.
# Clasificación final empleando un threshold de 0.8 para la clase 1.
# ==============================================================================
df_predicciones['clasificacion_custom_0.8'] = np.where(df_predicciones['1'] > 0.9, 1, 0)
df_predicciones.iloc[4:10, :]
| 0 | 1 | clasificacion_default_0.5 | clasificacion_custom_0.8 | |
|---|---|---|---|---|
| 4 | 0.793333 | 0.206667 | 0 | 0 |
| 5 | 0.606667 | 0.393333 | 0 | 0 |
| 6 | 0.186667 | 0.813333 | 1 | 0 |
| 7 | 0.360000 | 0.640000 | 1 | 0 |
| 8 | 0.126667 | 0.873333 | 1 | 0 |
| 9 | 0.160000 | 0.840000 | 1 | 0 |
¿Hasta que punto se debe de confiar en estas probabilidades?
Es muy importante tener en cuenta la diferencia entre la "visión" que tiene el modelo del mundo y el mundo real. Todo lo que sabe un modelo sobre el mundo real es lo que ha podido aprender de los datos de entrenamiento y, por lo tanto, tiene una "visión" limitada. Por ejemplo, supóngase que, en los datos de entrenamiento, todas las tiendas que están en zona urbana Urban='Yes' tienen ventas altas independientemente del valor que tomen el resto de predictores. Cuando el modelo trate de predecir una nueva observación, si esta está en zona urbana, clasificará a la tienda como ventas elevadas con un 100% de seguridad. Sin embargo, esto no significa que sea inequivocamente cierto, podría haber tiendas en zonas úrbanas que no tienen ventas elevadas pero, al no estar presentes en los datos de entrenamiento, el modelo no contempla esta posibilidad.
Teniendo en cuenta todo esto, hay que considerar las probabilidades generadas por el modelo como la seguridad que tiene este, desde su visión limitada, al realizar las predicciones. Pero no como la probailidad en el mundo real de que así lo sea.
Importancia de predictores¶
Pureza de nodos¶
importancia_predictores = pd.DataFrame(
{'predictor': X_train_prep.columns,
'importancia': modelo_final.feature_importances_}
)
print("Importancia de los predictores en el modelo")
print("-------------------------------------------")
importancia_predictores.sort_values('importancia', ascending=False)
Importancia de los predictores en el modelo -------------------------------------------
| predictor | importancia | |
|---|---|---|
| 9 | Price | 0.221835 |
| 5 | CompPrice | 0.115144 |
| 7 | Advertising | 0.114074 |
| 10 | Age | 0.100719 |
| 6 | Income | 0.097072 |
| 1 | ShelveLoc_Good | 0.086468 |
| 8 | Population | 0.074825 |
| 0 | ShelveLoc_Bad | 0.062682 |
| 11 | Education | 0.062288 |
| 2 | ShelveLoc_Medium | 0.027956 |
| 4 | US_Yes | 0.025334 |
| 3 | Urban_Yes | 0.011603 |
Permutación¶
importancia = permutation_importance(
estimator = modelo_final,
X = X_train_prep,
y = y_train,
n_repeats = 5,
scoring = 'neg_root_mean_squared_error',
n_jobs = cpu_count() - 1,
random_state = 123,
)
# Se almacenan los resultados (media y desviación) en un dataframe
df_importancia = pd.DataFrame(
{k: importancia[k] for k in ['importances_mean', 'importances_std']}
)
df_importancia['feature'] = X_train_prep.columns
df_importancia.sort_values('importances_mean', ascending=False)
| importances_mean | importances_std | feature | |
|---|---|---|---|
| 9 | 0.450240 | 0.024852 | Price |
| 1 | 0.343318 | 0.011515 | ShelveLoc_Good |
| 7 | 0.294198 | 0.010686 | Advertising |
| 5 | 0.258743 | 0.019633 | CompPrice |
| 0 | 0.253407 | 0.021248 | ShelveLoc_Bad |
| 10 | 0.174773 | 0.011008 | Age |
| 6 | 0.138299 | 0.014376 | Income |
| 8 | 0.072084 | 0.011716 | Population |
| 11 | 0.060537 | 0.031654 | Education |
| 4 | 0.034641 | 0.028284 | US_Yes |
| 3 | 0.000000 | 0.000000 | Urban_Yes |
| 2 | 0.000000 | 0.000000 | ShelveLoc_Medium |
# Gráfico
# ==============================================================================
fig, ax = plt.subplots(figsize=(3.5, 4))
df_importancia = df_importancia.sort_values('importances_mean', ascending=True)
ax.barh(
df_importancia['feature'],
df_importancia['importances_mean'],
xerr=df_importancia['importances_std'],
align='center',
alpha=0
)
ax.plot(
df_importancia['importances_mean'],
df_importancia['feature'],
marker="D",
linestyle="",
alpha=0.8,
color="r"
)
ax.set_title('Importancia de los predictores (train)')
ax.set_xlabel('Incremento del error tras la permutación');

Ambas estrategias identifican Price, Advertising y ShelveLoc como los predictores más influyentes, acorde a los datos de entrenamiento.
Random Forest con XGBoost¶
XGBoost se utiliza principalmente para crear modelos de tipo gradient boosting, sin embargo, dado que los modelos de random forest utilizan la misma representación de árboles y la misma estrategia de predicción, es posible configurar XGBoost para que utilice el algoritmo de random forest durante el entrenamiento. La configuración de los parámetros para entrenar modelos random forest es:
boosterdebe ser engbtree.subsampledebe ser en un valor menor que 1 para permitir la selección aleatoria de observaciones de entrenamiento (filas) en cada árbol.Uno de los parámetros
colsample_by*debe establecerse en un valor menor que 1 para permitir la selección aleatoria de columnas. Normalmente se utilizacolsample_bynodepara seleccionar columnas de manera aleatoria en cada división de árbol.num_parallel_treeindica el número de árboles que forman el modelo.num_boost_rounddebe establecerse en 1 para evitar que XGBoost combine múltiples modelos random forest. Este es un argumento del métodotrain(), y no de la inicialización.eta(alias:learning_rate) debe ser 1.
Para facilitar el uso de modelos random forest con XGBoost, existen dos clases XGBRFClassifier y XGBRFRegressor. Se trata de versiones de XGBClassifier y XGBRegressor que ya tienen por defecto el valor de los parámetros que consigue el entrenamiento de random forest en lugar de gradient boosting.
n_estimatorsespecifica el número de árboles. Internamente se convierte ennum_parallel_tree.learning_ratese establece en 1 de forma predeterminada.colsample_bynodeysubsamplese establecen en 0.8 de forma predeterminada.boosteres siempregbtree.
✎ Note
El principal beneficio de utilizar la librería XGBoost para entrenar modelos random forest frente a la implementación nativa de scikit-learn es la velocidad junto con su capacidad para manejar valores faltantes y variables categóricas.⚠ Warning
La implementación de XGBoost tiene algunas diferencias con respecto a la implementación de scikit-learn que pueden llevar a resultados diferentes.- XGBoost utiliza una aproximación de segundo orden a la función objetivo. Esto puede llevar a resultados que difieren de una implementación que utiliza el valor exacto de la función objetivo.
- XGBoost no realiza reemplazos al seleccionar observaciones de entrenamiento. Cada valor de entrenamiento puede aparecer como máximo una vez en cada muestreo.
from xgboost import XGBRFRegressor
import xgboost
print(f"Versión de XGBoost: {xgboost.__version__}")
# Descarga de datos
# ==============================================================================
url = (
'https://raw.githubusercontent.com/JoaquinAmatRodrigo/Estadistica-machine-learning-python/'
'master/data/Boston.csv'
)
datos = pd.read_csv(url, sep=',')
datos.head(3)
# División de los datos en entrenamiento y test
# ==============================================================================
X_train, X_test, y_train, y_test = train_test_split(
datos.drop(columns = "MEDV"),
datos['MEDV'],
random_state = 123
)
# Creación del modelo
# ==============================================================================
modelo = XGBRFRegressor(
n_estimators = 100,
tree_method = 'hist',
max_depth = 10,
max_leaves = 5,
learning_rate = 1.0,
subsample = 0.8,
colsample_bynode = 0.8,
reg_lambda = 1e-05,
reg_alpha = 0,
)
# Entrenamiento del modelo
# ==============================================================================
modelo.fit(X_train, y_train)
Versión de XGBoost: 3.0.0
XGBRFRegressor(base_score=None, booster=None, callbacks=None,
colsample_bylevel=None, colsample_bytree=None, device=None,
early_stopping_rounds=None, enable_categorical=False,
eval_metric=None, feature_types=None, feature_weights=None,
gamma=None, grow_policy=None, importance_type=None,
interaction_constraints=None, max_bin=None,
max_cat_threshold=None, max_cat_to_onehot=None,
max_delta_step=None, max_depth=10, max_leaves=5,
min_child_weight=None, missing=nan, monotone_constraints=None,
multi_strategy=None, n_estimators=100, n_jobs=None,
num_parallel_tree=None, objective='reg:squarederror',
random_state=None, ...)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Parameters
| learning_rate | 1.0 | |
| subsample | 0.8 | |
| colsample_bynode | 0.8 | |
| reg_lambda | 1e-05 | |
| objective | 'reg:squarederror' | |
| base_score | None | |
| booster | None | |
| callbacks | None | |
| colsample_bylevel | None | |
| colsample_bytree | None | |
| device | None | |
| early_stopping_rounds | None | |
| enable_categorical | False | |
| eval_metric | None | |
| feature_types | None | |
| feature_weights | None | |
| gamma | None | |
| grow_policy | None | |
| importance_type | None | |
| interaction_constraints | None | |
| max_bin | None | |
| max_cat_threshold | None | |
| max_cat_to_onehot | None | |
| max_delta_step | None | |
| max_depth | 10 | |
| max_leaves | 5 | |
| min_child_weight | None | |
| missing | nan | |
| monotone_constraints | None | |
| multi_strategy | None | |
| n_estimators | 100 | |
| n_jobs | None | |
| num_parallel_tree | None | |
| random_state | None | |
| reg_alpha | 0 | |
| sampling_method | None | |
| scale_pos_weight | None | |
| tree_method | 'hist' | |
| validate_parameters | None | |
| verbosity | None |
# Predicciones
# ==============================================================================
predicciones = modelo.predict(X_test)
# Error de test
# ==============================================================================
rmse = root_mean_squared_error(y_true=y_test, y_pred=predicciones)
print(f"El error (rmse) de test es: {rmse}")
El error (rmse) de test es: 4.759722741275837
Random Forest con LightGBM¶
Al igual que ocurre con XGBoost, los LightGBM también pueden configurarse para entrenar modelos random forest en lugar de gradient boosting.
from lightgbm import LGBMRegressor
import lightgbm
print(f"Versión de LightGBM: {lightgbm.__version__}")
# División de los datos en entrenamiento y test
# ==============================================================================
X_train, X_test, y_train, y_test = train_test_split(
datos.drop(columns = "MEDV"),
datos['MEDV'],
random_state = 123
)
# Creación del modelo
# ==============================================================================
modelo = LGBMRegressor(
boosting_type="rf",
n_estimators=50,
max_depth=10,
colsample_bytree=0.8,
subsample=0.8,
subsample_freq=1,
reg_alpha= 1, # L1 regularización
reg_lambda= 1, # L2 regularización
verbose=-1
)
# Entrenamiento del modelo
# ==============================================================================
modelo.fit(X_train, y_train)
Versión de LightGBM: 4.6.0
LGBMRegressor(boosting_type='rf', colsample_bytree=0.8, max_depth=10,
n_estimators=50, reg_alpha=1, reg_lambda=1, subsample=0.8,
subsample_freq=1, verbose=-1)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Parameters
| boosting_type | 'rf' | |
| num_leaves | 31 | |
| max_depth | 10 | |
| learning_rate | 0.1 | |
| n_estimators | 50 | |
| subsample_for_bin | 200000 | |
| objective | None | |
| class_weight | None | |
| min_split_gain | 0.0 | |
| min_child_weight | 0.001 | |
| min_child_samples | 20 | |
| subsample | 0.8 | |
| subsample_freq | 1 | |
| colsample_bytree | 0.8 | |
| reg_alpha | 1 | |
| reg_lambda | 1 | |
| random_state | None | |
| n_jobs | None | |
| importance_type | 'split' | |
| verbose | -1 |
# Predicciones
# ==============================================================================
predicciones = modelo.predict(X_test)
predicciones
# Error de test
# ==============================================================================
rmse = root_mean_squared_error(y_true=y_test, y_pred=predicciones)
print(f"El error (rmse) de test es: {rmse}")
El error (rmse) de test es: 4.8841999147856745
Extrapolación con Random Forest¶
Una límitación importante de los árboles de regresióna, y por lo tanto de Random Forest es que no extrapolan fuera del rango de entrenamiento. Cuando se aplica el modelo a una nueva observación, cuyo valor o valores de los predictores son superiores o inferiores a los observados en el entrenamiento, la predicción siempre es la media del nodo más cercano, independientemente de cuanto se aleje el valor. Vease el siguiente ejemplo en el que se entrenan dos modelos, un modelo lineal y un arbol de regresión, y luego se predicen valores de $X$ fuera del rango de entrenamiento.
import matplotlib.pyplot as plt
import numpy as np
from sklearn.ensemble import RandomForestRegressor
from sklearn.linear_model import LinearRegression
# Datos simulados
# ==============================================================================
X = np.linspace(0, 150, 100)
y = (X + 100) + np.random.normal(loc=0.0, scale=5.0, size=X.shape)
X_train = X[(X>=50) & (X<100)]
y_train = y[(X>=50) & (X<100)]
X_test_inf = X[X < 50]
y_test_inf = y[X < 50]
X_test_sup = X[X >= 100]
y_test_sup = y[X >= 100]
fig, ax = plt.subplots(figsize=(5, 3))
ax.plot(X_train, y_train, c='black', linestyle='dashed', label = "Datos train")
ax.axvspan(50, 100, color='gray', alpha=0.2, lw=0)
ax.plot(X_test_inf, y_test_inf, c='blue', linestyle='dashed', label = "Datos test")
ax.plot(X_test_sup, y_test_sup, c='blue', linestyle='dashed')
ax.set_title("Datos de entrenamiento y test")
plt.legend();

# Modelo lineal
modelo_lineal = LinearRegression()
modelo_lineal.fit(X_train.reshape(-1, 1), y_train)
# Modelo random forest
modelo_rf = RandomForestRegressor()
modelo_rf.fit(X_train.reshape(-1, 1), y_train)
# Predicciones
prediccion_lineal = modelo_lineal.predict(X.reshape(-1, 1))
prediccion_rf = modelo_rf.predict(X.reshape(-1, 1))
fig, ax = plt.subplots(figsize=(5, 3))
ax.plot(X_train, y_train, c='black', linestyle='dashed', label = "Datos train")
ax.axvspan(50, 100, color='gray', alpha=0.2, lw=0)
ax.plot(X_test_inf, y_test_inf, c='blue', linestyle='dashed', label = "Datos test")
ax.plot(X_test_sup, y_test_sup, c='blue', linestyle='dashed')
ax.plot(X, prediccion_lineal, c='firebrick',
label = "Prediccion modelo lineal")
ax.plot(X, prediccion_rf, c='orange',
label = "Prediccion modelo rf")
ax.set_title("Extrapolación de un modelo lineal y un modelo de random forest")
plt.legend();

La gráfica muestra que, con el modelo Random Forest, el valor predicho fuera del rango de entrenamiento, es el mismo independientemente de cuanto se aleje X. Una estrategia que permite que los modelos basados en árboles extrapolen es ajustar un modelo lineal en cada nodo terminal, pero esto no está disponible en Scikit-learn.
One-hot-encoding en Random Forest¶
Los modelos basados en árboles de decisión, entre ellos Random Forest, son capaces de utilizar predictores categóricos en su forma natural sin necesidad de convertirlos en variables dummy mediante one-hot-encoding. Sin embargo, en la práctica, depende de la implementación que tenga la librería o software utilizado. Esto tiene impacto directo en la estructura de los árboles generados y, en consecuencia, en los resultados predictivos del modelo y en la importancia calculada para los predictores.
El libro Feature Engineering and Selection A Practical Approach for Predictive Models By Max Kuhn, Kjell Johnson, los autores realizan un experimento en el que se comparan modelos entrenados con y sin one-hot-encoding en 5 sets de datos distintos, los resultados muestran que:
No hay una diferencia clara en términos de capacidad predictiva de los modelos. Solo en el caso de stocastic gradient boosting se apecian ligeras diferencias en contra de aplicar one-hot-encoding.
El entrenamiento de los modelos tarda en promedio 2.5 veces más cuando se aplica one-hot-encoding. Esto es debido al aumento de dimensionalidad al crear las nuevas variables dummy, obligando a que el algoritmo tenga que analizar muchos más puntos de división.
En base a los resultados obtenidos, los autores recomiendan no aplicar one-hot-encoding. Solo si los resultados son buenos, probar entonces si se mejoran al crear las variables dummy.
Otra comparación detallada puede encontrarse en Are categorical variables getting lost in your random forests? By Nick Dingwall, Chris Potts, donde los resultados muestran que:
Al convertir una variable categórica en múltiples variables dummy su importancia queda diluida, dificultando que el modelo pueda aprender de ella y perdiendo así capacidad predictiva. Este efecto es mayor cuantos más niveles tiene la variable original.
Al diluir la importancia de los predictores categóricos, estos tienen menos probabilidad de ser seleccionada por el modelo, lo que desvirtúa las métricas que miden la importancia de los predictores.
Por el momento, en scikit-learn es necesario hacer one-hot-encoding para convertir las variables categóricas en variables dummy. Las implementación de XGBoost y LightGBM sí permite utilizar directamente variables categóricas.
Extremely randomized trees¶
El algoritmo extremely randomized trees lleva la decorrelación de los árboles un paso más allá que random forest. Al igual que en random forest, en cada división de los nodos, solo se evalúa un subconjunto aleatorio de los predictores disponibles. Además de esto, en extremely randomized trees, dentro de cada predictor seleccionado solo se evalúa un subconjunto aleatorio de los posibles puntos de corte. En determinados escenarios, este método consigue reducir un poco más la varianza mejorando la capacidad predictiva del modelo. Puede encontrase este algoritmo en la case ExtraTrees de scikit-learn.
Comparación Random Forest y Gradient Boosting¶
Random Forest se compara con frecuencia con otro algoritmo basado en árboles, Gradient Boosting Trees. Ambos métodos son muy potentes y la superioridad de uno u otro depende del problema al que se apliquen. Algunos aspectos a tener en cuenta son:
Gracias al out-of-bag error, el método de Random Forest no necesita recurrir a validación cruzada para la optimización de sus hiperparámetros. Esto puede ser una ventaja muy importante cuando los requerimientos computacionales son limitantes. Esta característica también está presente en Stochastic Gradient Boosting pero no en AdaBoost y Gradient Boosting.
Random forest tiene menos hiperparámetros, lo que hace más sencillo su correcta implementación.
Si existe una proporción alta de predictores irrelevantes, Random Forest puede verse perjudicado, sobre todo a medida que se reduce el número de predictores ($m$) evaluados. Supóngase que, de 100 predictores, 90 de ellos no aportan información (son ruidos). Si el hiperparámetro $m$ es igual a 3 (en cada división se evalúan 3 predictores aleatorios), es muy probable que los 3 predictores seleccionados sean de los que no aportan nada. Aun así, el algoritmo seleccionará el mejor de ellos, incorporándolo al modelo. Cuanto mayor sea el porcentaje de predictores no relevantes, mayor la frecuencia con la que esto ocurre, por lo que los árboles que forman el Random Forest contendrán predictores irrelevantes. Como consecuencia, se reduce su capacidad predictiva. En el caso de Gradient Boosting Trees, siempre se evalúan todos los predictores, por lo que los no relevantes se ignoran.
En Random Forest, cada modelo que forma el ensemble es independiente del resto, esto permite que el entrenamiento sea paralelizable.
Random Forest no sufre problemas de overfitting por muchos árboles que se agreguen.
Si se realiza una buena optimización de hiperparámetros, Gradient Boosting Trees suele obtener resultados ligeramente superiores.
Información de sesión¶
import session_info
session_info.show(html=False)
----- joblib 1.4.2 lightgbm 4.6.0 matplotlib 3.10.1 numpy 2.2.6 optuna 3.6.2 pandas 2.3.1 session_info v1.0.1 sklearn 1.7.1 statsmodels 0.14.4 xgboost 3.0.0 ----- IPython 9.1.0 jupyter_client 8.6.3 jupyter_core 5.7.2 jupyterlab 4.4.5 notebook 7.4.5 ----- Python 3.12.9 | packaged by Anaconda, Inc. | (main, Feb 6 2025, 18:56:27) [GCC 11.2.0] Linux-6.14.0-27-generic-x86_64-with-glibc2.39 ----- Session information updated at 2025-08-18 11:52
Bibliografía¶
Introduction to Statistical Learning, Gareth James, Daniela Witten, Trevor Hastie and Robert Tibshirani
Applied Predictive Modeling Max Kuhn, Kjell Johnson
T.Hastie, R.Tibshirani, J.Friedman. The Elements of Statistical Learning. Springer, 2009
Bradley Efron and Trevor Hastie. 2016. Computer Age Statistical Inference: Algorithms, Evidence, and Data Science (1st. ed.). Cambridge University Press, USA.
Instrucciones para citar¶
¿Cómo citar este documento?
Si utilizas este documento o alguna parte de él, te agradecemos que lo cites. ¡Muchas gracias!
Random Forest con Python por Joaquín Amat Rodrigo, disponible bajo una licencia Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0 DEED) en https://www.cienciadedatos.net/documentos/py08_random_forest_python.html
¿Te ha gustado el artículo? Tu ayuda es importante
Tu contribución me ayudará a seguir generando contenido divulgativo gratuito. ¡Muchísimas gracias! 😊



Este documento creado por Joaquín Amat Rodrigo tiene licencia Attribution-NonCommercial-ShareAlike 4.0 International.
Se permite:
-
Compartir: copiar y redistribuir el material en cualquier medio o formato.
-
Adaptar: remezclar, transformar y crear a partir del material.
Bajo los siguientes términos:
-
Atribución: Debes otorgar el crédito adecuado, proporcionar un enlace a la licencia e indicar si se realizaron cambios. Puedes hacerlo de cualquier manera razonable, pero no de una forma que sugiera que el licenciante te respalda o respalda tu uso.
-
No-Comercial: No puedes utilizar el material para fines comerciales.
-
Compartir-Igual: Si remezclas, transformas o creas a partir del material, debes distribuir tus contribuciones bajo la misma licencia que el original.